Dimanganese decacarbonyl
Dimanganese decacarbonyl,[3] which has the chemical formula Mn2(CO)10, is a binary bimetallic carbonyl complex centered around the first row transition metal manganese.
The first reported synthesis of Mn2(CO)10 was in 1954 at Linde Air Products Company and was performed by Brimm, Lynch, and Sesny.
Since its first synthesis, Mn2(CO)10 has been use sparingly as a reagent in the synthesis of other chemical species, but has found the most use as a simple system on which to study fundamental chemical and physical phenomena, most notably, the metal-metal bond.
Dimanganese decacarbonyl is also used as a classic example to reinforce fundamental topics in organometallic chemistry like d-electron count, the 18-electron rule, oxidation state, valency,[5] and the isolobal analogy.
Many procedures have been reported for the synthesis of Mn2(CO)10 since 1954, the two most common general types are discussed herein.
As previously mentioned, Mn2(CO)10 was first prepared in 1954 by Brimm, Lynch, and Sesny, albeit in yields of ~1%, by the reduction of manganese(II) iodide with magnesium(0) under 3000 psi (~200 atm) of carbon monoxide (CO).
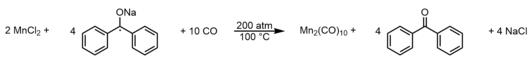
A more efficient preparation was developed in 1958 and entails reduction of anhydrous manganese(II) chloride with sodium benzophenone ketyl radical under similarly high pressures (200 atm) of CO.[6] This method yielded ~32% of the dimanganese decacarbonyl complex, producing enough material for the first real opportunities to rigorously study the chemical and physical properties of the molecule.
This method is represented by the balanced equation: Despite successes in the synthesis of Mn2(CO)10, the safety concerns and limited batch size surrounding high pressure carbonylation methods necessitated alternative, low pressure procedures to obtain the target compound.
In 1968, the first ambient CO pressure carbonylation synthesis of Mn2(CO)10 was reported from the commercially available and inexpensive methylcyclopentadienyl manganese tricarbonyl (MMT) and sodium(0) as the reductant.
The efficiency of the method ranged from 16 to 20% yield, lower than what was previously reported, however, it could be performed more safely and on mole scale.
These preparations differ, however, by beginning with manganese precursors, sometimes commercially available, that need no additional CO ligands and simply dimerize to form the target molecule.
This poses the significant logistic and safety advantage of not dealing with toxic CO gas and is the prevailing general method for the academic synthesis of Mn2(CO)10.
The first explicit success was published in 1977, and featured a pentacarbonylhydridomanganese(-I) Mn source, oxidized by Se(PF2)2:[8]
Other terminal oxidants achieve the same effect,[9][10][11] and stable pentacarbonylmanganate (Mn(CO)−5) salts can substitute for the hydride.
[12][13][14] Thus for example triphenylcyclopropenium tetrafluoroborate reacts with sodium pentacarbonyl manganate to produce the dimer of each:[15] Similar methods exist for Mn(CO)5X compounds where X = Cl, Br, or I; and, more rarely, for Mn(CO)+6 bound with a weakly coordinating anion.
In this instance, manganese is both the oxidant and reductant, producing two formal Mn(0) atoms:[22]
High precision crystallographic and theoretical studies of the physical and electronic structures of Mn2(CO)10 have been performed and are discussed with respect to the published literature below, however, a qualitative approach can also be taken to predict its constitutional structure using fundamental principles of inorganic and organometallic chemistry.
The stoichiometric composition of Mn2(CO)10, derived from elemental analysis, informs a 5:1 ratio of CO to Mn.
This is a highly unstable configuration, isolobal to the methyl radical, which can be expected to homodimerize to the constitutionally symmetric dinuclear complex in order for both Mn nuclei to achieve an 18-electron, noble gas configuration.
This hypothesized structure was confirmed explicitly through x-ray diffraction studies, first in two dimensions in 1957,[23] followed by its single crystal three-dimensional analysis in 1963.
[24] The crystal structure of Mn2(CO)10 was redetermined at high precision at room temperature in 1981 and bond lengths mentioned herein refer to results from that study.
Initial fundamental experimental and theoretical studies on the electronic structure of Mn2(CO)10 were performed used a mixture of photoelectron spectroscopy, infrared spectroscopy, and an iterative extended-Hückel-type molecular orbital calculation.
[29] The electronic structure described herein, along with relevant orbital plots, are reproduced from the methods used in that study using Orca (5.0.3)[30] and visualized using IBOView (v20150427).
The pi-backbonding interactions illustrated below occur between the t2g d-orbital set and the CO π* antibonding orbitals.
The degenerate dxz and dyz backbonding interactions with both axial and equatorial CO ligands is the HOMO-15.
Other large contributions made in this area were by Ahmed Zewail using ultrafast, femtosecond spectroscopy en route to his 1999 Nobel Prize.
[32] His discoveries elucidated much about the time scales and energies associated with the molecular motions of Mn2(CO)10, as well as the Mn-Mn and Mn-C bond cleavage events.
[33] Mn2(CO)10 is air stable as a crystalline solid, but solutions require Schlenk techniques.
Redox neutral cleavage is possible both thermally and photochemically, producing two equivalents of the Mn(0) radical.
Selective mono-oxidation of the Mn-Mn bond is most often done via addition of classical metal oxidants (e.g. CeIV, PbIV, etc) or weak homonuclear single covalent bonds of the form X-X (X is group 16 or 17 element).