p53
[17] A study of Arab women found that proline homozygosity at TP53 codon 72 is associated with a decreased risk for breast cancer.
[22] A 2011 study of a Brazilian birth cohort found an association between the non-mutant arginine TP53 and individuals without a family history of cancer.
[23] Another 2011 study found that the p53 homozygous (Pro/Pro) genotype was associated with a significantly increased risk for renal cell carcinoma.
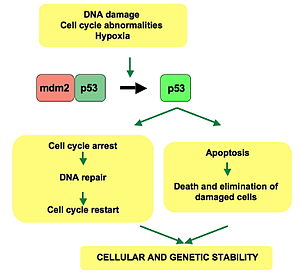
[24] p53 plays a role in regulation or progression through the cell cycle, apoptosis, and genomic stability by means of several mechanisms: WAF1/CIP1 encodes for p21 and hundreds of other down-stream genes.
A mutant p53 will no longer bind DNA in an effective way, and, as a consequence, the p21 protein will not be available to act as the "stop signal" for cell division.
[29][30] Levels of p53 play an important role in the maintenance of stem cells throughout development and the rest of human life.
Work in mouse embryonic stem cells has recently shown however that the expression of P53 does not necessarily lead to differentiation.
[40] As tumors grow they need to recruit new blood vessels to supply them, and p53 inhibits that by (i) interfering with regulators of tumor hypoxia that also affect angiogenesis, such as HIF1 and HIF2, (ii) inhibiting the production of angiogenic promoting factors, and (iii) directly increasing the production of angiogenesis inhibitors, such as arresten.
[41][42] p53 by regulating Leukemia Inhibitory Factor has been shown to facilitate implantation in the mouse and possibly human reproduction.
[45] p53 is activated in response to myriad stressors – including DNA damage (induced by either UV, IR, or chemical agents such as hydrogen peroxide), oxidative stress,[46] osmotic shock, ribonucleotide depletion, viral lung infections[47] and deregulated oncogene expression.
The N-terminal transcriptional activation domain contains a large number of phosphorylation sites and can be considered as the primary target for protein kinases transducing stress signals.
[citation needed] The protein kinases that are known to target this transcriptional activation domain of p53 can be roughly divided into two groups.
A second group of protein kinases (ATR, ATM, CHK1 and CHK2, DNA-PK, CAK, TP53RK) is implicated in the genome integrity checkpoint, a molecular cascade that detects and responds to several forms of DNA damage caused by genotoxic stress.
People who inherit only one functional copy of the TP53 gene will most likely develop tumors in early adulthood, a disorder known as Li–Fraumeni syndrome.
[citation needed] The TP53 gene can also be modified by mutagens (chemicals, radiation, or viruses), increasing the likelihood for uncontrolled cell division.
For example, restoration of endogenous p53 function in lymphomas may induce apoptosis, while cell growth may be reduced to normal levels.
[57][58] The first commercial gene therapy, Gendicine, was approved in China in 2003 for the treatment of head and neck squamous cell carcinoma.
Persistent infection of the cervix over the years can cause irreversible changes leading to carcinoma in situ and eventually invasive cervical cancer.
[60] The p53 protein is continually produced and degraded in cells of healthy people, resulting in damped oscillation (see a stochastic model of this process in [61]).
[62] This image shows different patterns of p53 expression in endometrial cancers on chromogenic immunohistochemistry, whereof all except wild-type are variably termed abnormal/aberrant/mutation-type and are strongly predictive of an underlying TP53 mutation:[63] Suppression of p53 in human breast cancer cells is shown to lead to increased CXCR5 chemokine receptor gene expression and activated cell migration in response to chemokine CXCL13.
[66] One study found that p53 and Myc proteins were key to the survival of Chronic Myeloid Leukaemia (CML) cells.
[75][76] Mathematical models also indicate that the p53 concentration oscillates much faster once teratogens, such as double-stranded breaks (DSB) or UV radiation, are introduced to the system.
This supports and models the current understanding of p53 dynamics, where DNA damage induces p53 activation (see p53 regulation for more information).
Its role as a tumor suppressor gene was revealed in 1989 by Bert Vogelstein at the Johns Hopkins School of Medicine and Arnold Levine at Princeton University.
[81][82] p53 went on to be identified as a transcription factor by Guillermina Lozano working at MD Anderson Cancer Center.
[83] Warren Maltzman, of the Waksman Institute of Rutgers University first demonstrated that TP53 was responsive to DNA damage in the form of ultraviolet radiation.
[84] In a series of publications in 1991–92, Michael Kastan of Johns Hopkins University, reported that TP53 was a critical part of a signal transduction pathway that helped cells respond to DNA damage.
Most of these mutations destroy the ability of the protein to bind to its target DNA sequences, and thus prevents transcriptional activation of these genes.
This difference is due to the high number of proline residues in the protein, which slow its migration on SDS-PAGE, thus making it appear heavier than it actually is.
Furthermore, the usage of an internal promoter in intron 4 causes the ∆133 and ∆160 isoforms, which lack the TAD domain and a part of the DBD.