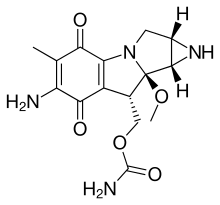
Aziridines
Aziridine is less basic than acyclic aliphatic amines, with a pKa of 7.9 for the conjugate acid, due to increased s character of the nitrogen free electron pair.
An amine functional group displaces the adjacent halide in an intramolecular nucleophilic substitution reaction to generate an aziridine.
In the Blum-Ittah aziridine synthesis, sodium azide opens an epoxide, followed by reduction of the azide with triphenylphosphine accompanied by expulsion of nitrogen gas:[8] Another method involves the ring-opening reaction of an epoxide with amines, followed by ring closing with the Mitsunobu reaction.
[10] Aziridines are obtained by treating a mono-, di-, tri- or tetra-substituted alkene (olefin) with O-(2,4-dinitrophenyl)hydroxylamine [de] (DPH) in the presence of rhodium catalysts: For instance, 2-phenyl-3-methylaziridine can be synthesized by this method and then converted by ring opening reaction to (D)- and (L)-amphetamine (the two active ingredients in Adderall).
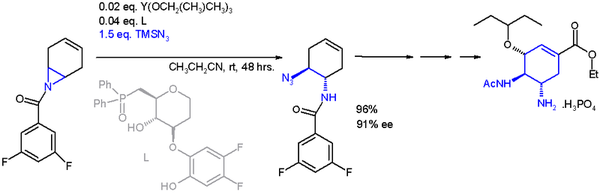
[16][17] One application of a ring-opening reaction in asymmetric synthesis is that of trimethylsilylazide TMSN3 with an asymmetric ligand[18] in scheme 2[19] in an organic synthesis of oseltamivir: Certain N-substituted azirines with electron withdrawing groups on both carbons form azomethine ylides in an electrocyclic thermal or photochemical ring-opening reaction.
Lewis acids, such as B(C6F5)3, can induce decomposition of the ring to a carbocation and linear azanide, which then attack unsaturated moieties in tandem.
[29] In making the overall evaluation, the IARC Working Group took into consideration that aziridine is a direct-acting alkylating agent, which is mutagenic in a wide range of test systems and forms DNA adducts that are promutagenic.