Crystal field theory
CFT successfully accounts for some magnetic properties, colors, hydration enthalpies, and spinel structures of transition metal complexes, but it does not attempt to describe bonding.
The theory is developed by considering energy changes of the five degenerate d-orbitals upon being surrounded by an array of point charges consisting of the ligands.
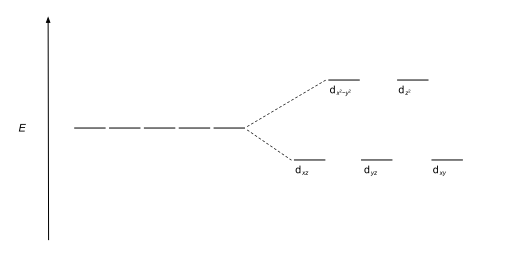
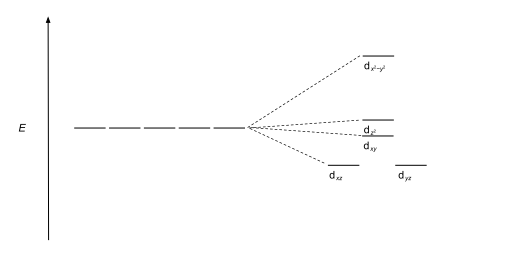
This splitting is affected by the following factors: The most common type of complex is octahedral, in which six ligands form the vertices of an octahedron around the metal ion.
These labels are based on the theory of molecular symmetry: they are the names of irreducible representations of the octahedral point group, Oh.
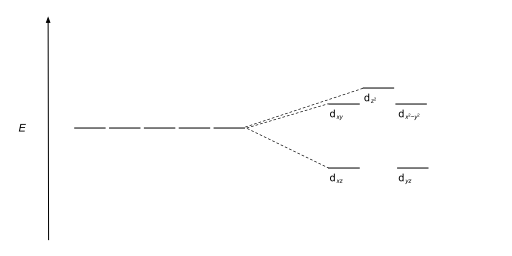
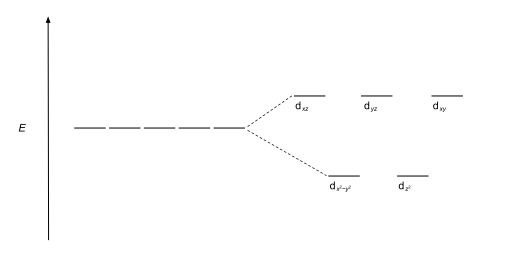
Furthermore, since the ligand electrons in tetrahedral symmetry are not oriented directly towards the d-orbitals, the energy splitting will be lower than in the octahedral case.
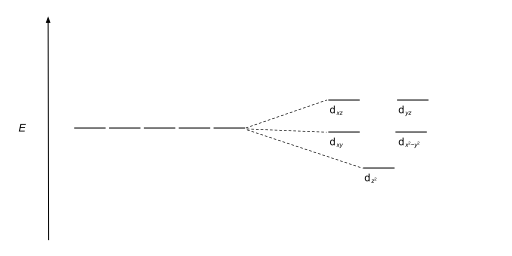
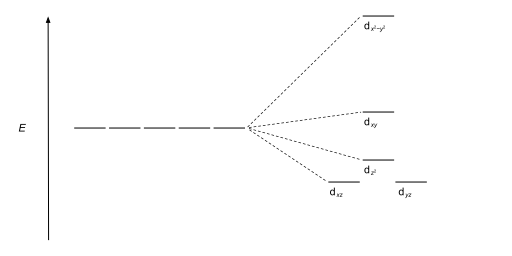
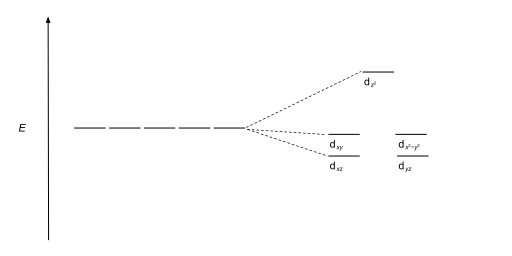
The size of the gap Δ between the two or more sets of orbitals depends on several factors, including the ligands and geometry of the complex.
The oxidation state of the metal also contributes to the size of Δ between the high and low energy levels.
Therefore, the lower energy orbitals are completely filled before population of the upper sets starts according to the Aufbau principle.
So, one electron is put into each of the five d-orbitals in accord with Hund's rule, and "high spin" complexes are formed before any pairing occurs.
As a result of this, if there are any electrons occupying these orbitals, the metal ion is more stable in the ligand field relative to the barycenter by an amount known as the CFSE.
Conversely, the eg orbitals (in the octahedral case) are higher in energy than in the barycenter, so putting electrons in these reduces the amount of CFSE.
The optical properties (details of absorption and emission spectra) of many coordination complexes can be explained by Crystal Field Theory.