DNA repair
[3] The 2015 Nobel Prize in Chemistry was awarded to Tomas Lindahl, Paul Modrich, and Aziz Sancar for their work on the molecular mechanisms of DNA repair processes.
These modifications can in turn disrupt the molecules' regular helical structure by introducing non-native chemical bonds or bulky adducts that do not fit in the standard double helix.
Constitutive (spontaneous) DNA damage caused by endogenous oxidants can be detected as a low level of histone H2AX phosphorylation in untreated cells.
Mitochondrial DNA (mtDNA) is located inside mitochondria organelles, exists in multiple copies, and is also tightly associated with a number of proteins to form a complex known as the nucleoid.
Senescence, an irreversible process in which the cell no longer divides, is a protective response to the shortening of the chromosome ends, called telomeres.
Depending on the type of damage inflicted on the DNA's double helical structure, a variety of repair strategies have evolved to restore lost information.
The photoreactivation process directly reverses this damage by the action of the enzyme photolyase, whose activation is obligately dependent on energy absorbed from blue/UV light (300–500 nm wavelength) to promote catalysis.
Partially overlapping fragments are then used for synthesis of homologous regions through a moving D-loop that can continue extension until complementary partner strands are found.
[39] Topoisomerases introduce both single- and double-strand breaks in the course of changing the DNA's state of supercoiling, which is especially common in regions near an open replication fork.
From a cellular perspective, risking the introduction of point mutations during translesion synthesis may be preferable to resorting to more drastic mechanisms of DNA repair, which may cause gross chromosomal aberrations or cell death.
Cells exposed to ionizing radiation, ultraviolet light or chemicals are prone to acquire multiple sites of bulky DNA lesions and double-strand breaks.
[46] The global response to damage is an act directed toward the cells' own preservation and triggers multiple pathways of macromolecular repair, lesion bypass, tolerance, or apoptosis.
[52] γH2AX (H2AX phosphorylated on serine 139) can be detected as soon as 20 seconds after irradiation of cells (with DNA double-strand break formation), and half maximum accumulation of γH2AX occurs in one minute.
The loss of LexA repressor induces transcription of the SOS genes and allows for further signal induction, inhibition of cell division and an increase in levels of proteins responsible for damage processing.
In other classes and phyla, the sequence of SOS boxes varies considerably, with different length and composition, but it is always highly conserved and one of the strongest short signals in the genome.
A large amount of damage to a cell leaves it with an important decision: undergo apoptosis and die, or survive at the cost of living with a modified genome.
[66] In similar manner, mice deficient in a key repair and transcription protein that unwinds DNA helices have premature onset of aging-related diseases and consequent shortening of lifespan.
Caloric restriction reproducibly results in extended lifespan in a variety of organisms, likely via nutrient sensing pathways and decreased metabolic rate.
[73] The mammalian homolog of SIR-2 is known to induce downstream DNA repair factors involved in NHEJ, an activity that is especially promoted under conditions of caloric restriction.
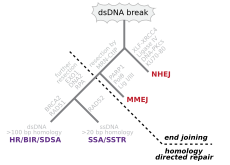
BRCA1 and BRCA2, two important genes whose mutations confer a hugely increased risk of breast cancer on carriers,[80] are both associated with a large number of DNA repair pathways, especially NHEJ and homologous recombination.
However, synthetic lethality therapeutic approaches have been questioned due to emerging evidence of acquired resistance, achieved through rewiring of DNA damage response pathways and reversion of previously inhibited defects.
[84] Previous studies have shown an elevated DNA damage response in cell-culture models with oncogene activation[85] and preneoplastic colon adenomas.
In experimental mouse models, loss of DNA damage response-mediated cell senescence was observed after using a short hairpin RNA (shRNA) to inhibit the double-strand break response kinase ataxia telangiectasia (ATM), leading to increased tumor size and invasiveness.
[91] Classically, cancer has been viewed as a set of diseases that are driven by progressive genetic abnormalities that include mutations in tumour-suppressor genes and oncogenes, and chromosomal aberrations.
[28] FEN1, the flap endonuclease in MMEJ, is epigenetically increased by promoter hypomethylation and is over-expressed in the majority of cancers of the breast,[119] prostate,[120] stomach,[121][122] neuroblastomas,[123] pancreas,[124] and lung.
One mechanism underlying this involves the histone modification H3K36me3, which can recruit mismatch repair proteins,[130] thereby lowering mutation rates in H3K36me3-marked regions.
[131] Another important mechanism concerns nucleotide excision repair, which can be recruited by the transcription machinery, lowering somatic mutation rates in active genes[129] and other open chromatin regions.
[146] Further work by Allen et al.[150] showed that NHEJ of a DNA double-strand break in a cell could give rise to some progeny cells having repressed expression of the gene harboring the initial double-strand break and some progeny having high expression of that gene due to epigenetic alterations associated with NHEJ repair.
[151] The ability of a large number of protein structural motifs to catalyze relevant chemical reactions has played a significant role in the elaboration of repair mechanisms during evolution.
On the other hand, DNA damage repair and protection does influence the rate of accumulation of irreparable, advantageous, code expanding, inheritable mutations, and slows down the evolutionary mechanism for expansion of the genome of organisms with new functionalities.