NMDA receptor
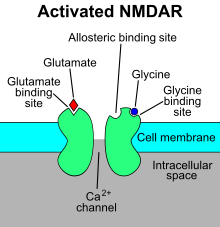
Activation of NMDA receptors results in the opening of the ion channel that is nonselective to cations, with a combined reversal potential near 0 mV.
[8][9][10][11] Ca2+ flux through NMDA receptors in particular is thought to be critical in synaptic plasticity, a cellular mechanism for learning and memory, due to proteins which bind to and are activated by Ca2+ ions.
[16][17][18][19] However, hypofunction of NMDA receptors (due to glutathione deficiency or other causes) may be involved in impairment of synaptic plasticity[20] and could have other negative repercussions.
Extrasynaptic NMDA receptors promote death signaling; they cause transcriptional shut-off, mitochondrial dysfunction, and structural disintegration.
[24][25] This pathological triad of extrasynaptic NMDA receptor signaling represents a common conversion point in the etiology of several acute and chronic neurodegenerative conditions.
[27] A fortuitous finding was made in 1968 when a woman was taking amantadine as flu medicine and experienced remarkable remission of her Parkinson's symptoms.
[15] Functional NMDA receptors are heterotetramers comprising different combinations of the GluN1, GluN2 (A-D), and GluN3 (A-B) subunits derived from distinct gene families (Grin1-Grin3).
This intricate molecular structure and genetic diversity enable the receptor to carry out a wide range of physiological functions within the nervous system.
[30][31] All the subunits share a common membrane topology that is dominated by a large extracellular N-terminus, a membrane region comprising three transmembrane segments, a re-entrant pore loop, an extracellular loop between the transmembrane segments that are structurally not well known, and an intracellular C-terminus, which are different in size depending on the subunit and provide multiple sites of interaction with many intracellular proteins.
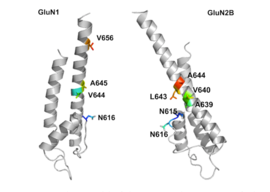
[34] The GluN2B subunit has been involved in modulating activity such as learning, memory, processing and feeding behaviors, as well as being implicated in number of human derangements.
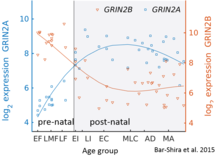
Multiple receptor isoforms with distinct brain distributions and functional properties arise by selective splicing of the GluN1 transcripts and differential expression of the GluN2 subunits.
[18] Memantine is an example of an uncompetitive channel blocker of the NMDA receptor, with a relatively rapid off-rate and low affinity.
In classic circuits, GluN2B is mainly present in immature neurons and in extrasynaptic locations such as growth cones,[38] and contains the binding-site for the selective inhibitor ifenprodil.
[39] However, in pyramidal cell synapses in the newly evolved primate dorsolateral prefrontal cortex, GluN2B are exclusively within the postsynaptic density, and mediate higher cognitive operations such as working memory.
[46] This may in part account for greater memory abilities in the immediate postnatal period compared to late in life, which is the principle behind genetically altered 'doogie mice'.
[53][56] In order to support the localization hypothesis, it would be necessary to show differing cellular signaling pathways are activated by NMDA receptors based on its location within the cell membrane.
In addition, both synaptic and extrasynaptic activity are involved in expressing a full LTD.[64] Another factor that seems to affect NMDAR induced toxicity is the observed variation in subunit makeup.
These side effects are, in part, observed because the NMDA receptors do not just signal for cell death but also play an important role in its vitality.
[73] D-serine, an antagonist/inverse co-agonist of t-NMDA receptors, which is made in the brain, has been shown to mitigate neuron loss in an animal model of temporal lobe epilepsy.
3,5-Dibromo-L-phenylalanine, a naturally occurring halogenated derivative of L-phenylalanine, is a weak partial NMDA receptor agonist acting on the glycine site.
[79] Other partial agonists of the NMDA receptor acting on novel sites such as rapastinel (GLYX-13) and apimostinel (NRX-1074) are now viewed for the development of new drugs with antidepressant and analgesic effects without obvious psychotomimetic activities.
[80] Positive allosteric modulators include: Antagonists of the NMDA receptor are used as anesthetics for animals and sometimes humans, and are often used as recreational drugs due to their hallucinogenic properties, in addition to their unique effects at elevated dosages such as dissociation.
When certain NMDA receptor antagonists are given to rodents in large doses, they can cause a form of brain damage called Olney's lesions.
So far, the published research on Olney's lesions is inconclusive in its occurrence upon human or monkey brain tissues with respect to an increase in the presence of NMDA receptor antagonists.
[98] Nitromemantine is a second-generation derivative of memantine, it reduces excitotoxicity mediated by overactivation of the glutamatergic system by blocking NMDA receptor without sacrificing safety.
[16] Uncompetitive NMDA receptor antagonists block within the ion channel at the Mg2+ site (pore region) and prevent excessive influx of Ca2+.
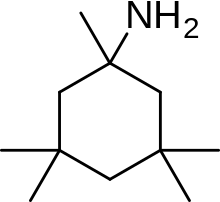
The methyl side groups play an important role in increasing the affinity to the open NMDA receptor channels and making it a much better neuroprotective drug than amantadine.
NMDA receptors are modulated by a number of endogenous and exogenous compounds and play a key role in a wide range of physiological (e.g., memory) and pathological processes (e.g., excitotoxicity).
[121][122] Research suggests that tianeptine produces antidepressant effects through indirect alteration and inhibition of glutamate receptor activity and release of BDNFTooltip brain-derived neurotrophic factor, in turn affecting neural plasticity.
NMDAR antagonists, for instance eliprodil, gavestinel, licostinel, and selfotel have been extensively investigated for the treatment of excitotoxicity-mediated neurotoxicity in situations like ischemic stroke and traumatic brain injury, but were unsuccessful in clinical trials used in small doses to avoid sedation, but NMDAR antagonists can block Spreading Depolarizations in animals and in patients with brain injury.