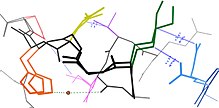
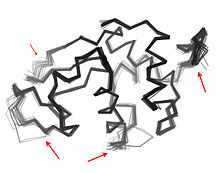
Protein dynamics
In molecular biology, proteins are generally thought to adopt unique structures determined by their amino acid sequences.
[11][12] When these coupled residues form pathways linking functionally important parts of a protein, they may participate in allosteric signaling.
A detailed analysis by Gerstein led to the classification of two basic types of domain motion; hinge and shear.
[25] A study by Hayward[26] found that the termini of α-helices and β-sheets form hinges in a large number of cases.
α-helices that preserve their hydrogen bonding network when bent are found to behave as mechanical hinges, storing `elastic energy' that drives the closure of domains for rapid capture of a substrate.
[citation needed] The analysis of the internal dynamics of structurally different, but functionally similar enzymes has highlighted a common relationship between the positioning of the active site and the two principal protein sub-domains.
In fact, for several members of the hydrolase superfamily, the catalytic site is located close to the interface separating the two principal quasi-rigid domains.
[14] Such positioning appears instrumental for maintaining the precise geometry of the active site, while allowing for an appreciable functionally oriented modulation of the flanking regions resulting from the relative motion of the two sub-domains.
[31] This argument suggests that proteins have evolved to have stable, mostly unique folded structures, but the unavoidable residual flexibility leads to some degree of functional promiscuity, which can be amplified/harnessed/diverted by subsequent mutations.
[32] However, there is growing awareness that intrinsically unstructured proteins are quite prevalent in eukaryotic genomes,[33] casting further doubt on the simplest interpretation of Anfinsen's dogma: "sequence determines structure (singular)".
In effect, the new paradigm is characterized by the addition of two caveats: "sequence and cellular environment determine structural ensemble".