Wender Taxol total synthesis
The taxol synthesis started from the terpene verbenone 1 in Scheme 1, which is the oxidation product of naturally occurring α-pinene and forming ring A.
Ozonolysis of the prenyl group (more electron-rich than the internal double bond) formed aldehyde 4, which, after isomerization or photorearrangement to the chrysanthenone 5, was reacted with the lithium salt (via LDA) of the ethyl ester of propiolic acid 6 in a nucleophilic addition to the alcohol 7.
The silyl ether protective group was removed by reaction with acetic acid to alcohol 11, which was then oxidized to the ketone 12 with RuCl2(PPh3)3 and NMO as the sacrificial catalyst.
The C1 position was next oxidized by the phosphite ester, P(OEt)3 and the strong base KOt-Bu, and oxygen to alcohol 20 (the stereochemistry controlled by bowl-shaped AB ring with hydroxylation from unhindered convex direction), the primary alcohol group was deprotected with ammonium chloride in methanol to diol 21 and two reductions first with NaBH4 to triol 22 and then hydrogen gas and Crabtree's catalyst give triol 23.
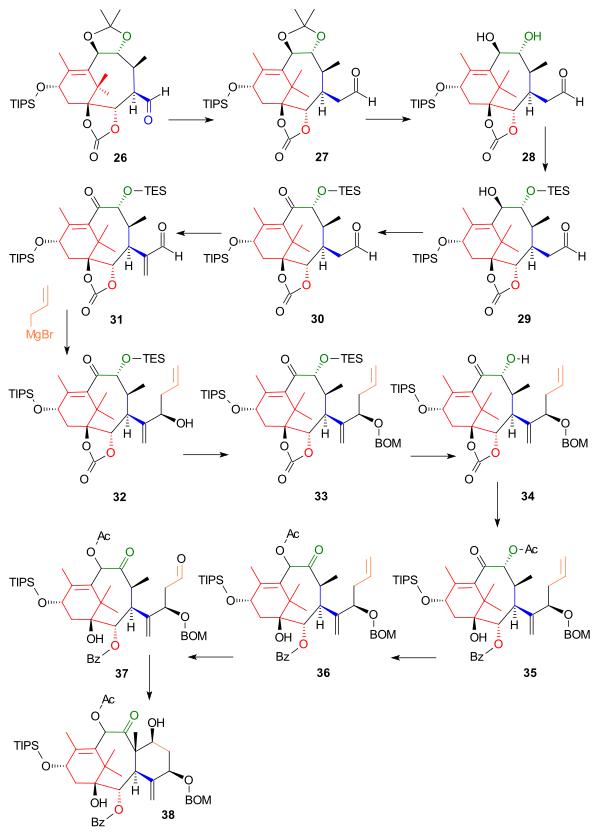
These positions were protected by trimethylsilyl chloride and pyridine to 24 and then triphosgene to 25 in order to facilitate the oxidation of the primary alcohol group to the aldehyde 26 by PCC.
Two additional countermeasures were required: reprotection of the diol as the carbonate ester 45 with triphosgene and removal of the benzoate group (KCN) to alcohol 46 in preparation of the actual ring closure to the oxetane 47 with N,N-diisopropylethylamine.