Wolff rearrangement
The Wolff rearrangement is a reaction in organic chemistry in which an α-diazocarbonyl compound is converted into a ketene by loss of dinitrogen with accompanying 1,2-rearrangement.
[3] The Wolff rearrangement has great synthetic utility due to the accessibility of α-diazocarbonyl compounds, variety of reactions from the ketene intermediate, and stereochemical retention of the migrating group.
[2] However, the Wolff rearrangement has limitations due to the highly reactive nature of α-diazocarbonyl compounds, which can undergo a variety of competing reactions.
[3] In this last case, the reaction is sensitive to the transition metal; silver (I) oxide or other Ag(I) catalysts work well and are generally used.
The Wolff rearrangement has been used in many total syntheses; the most common use is trapping the ketene intermediate with nucleophiles to form carboxylic acid derivatives.
In 1902, Wolff discovered that treating diazoacetophenone with silver (I) oxide and water resulted in formation of phenylacetic acid.
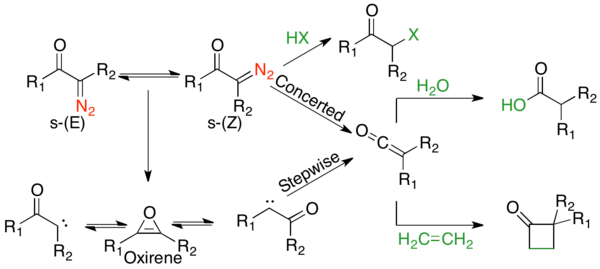
α-diazocarbonyl compounds are generally locally planar, with large rotational barriers (55–65 kJ/mol) due to C-C olefin character between the carbonyl and α-carbon, illustrated in the rightmost resonance structure.
[6] Such a large barrier slows molecular rotations sufficiently to lead to an equilibrium between two conformers, an s-trans and s-cis-conformer.
s-cis-Conformers are electronically favored due to Coulombic attraction between the oxygen with a partial negative charge and the cationic nitrogen, as seen in the rightmost resonance structure.
The first is that rate constants of Wolff rearrangements depend on the stability of the formed carbene, rather than the migratory aptitude of the migrating group.
[10] The most definitive evidence is isotopic scrambling of the ketene, as predicted by an oxirene intermediate, which can only occur in the stepwise path.
[12] α-diazocyclohexanone shows no label scrambling under photolytic conditions, as it is entirely s-cis, and thus all substrate goes through the concerted mechanism, avoiding the oxirene intermediate.
The observation that the migratory ability of a substituent is inversely proportional to amount of carbene formed, indicates that under photolysis, there are competing pathways for many Wolff reactions.
[1] However, for all substrates except cyclic α-diazo ketones that exist solely in the s-cis conformation, products come from a combination of both pathways.
The primary ways to prepare these substrates today are via the Arndt-Eistert procedure, the Franzen modification to the Dakin-West reaction, and diazo-transfer methods.
These α-diazo ketones are unstable under acidic conditions, as the α-carbon can be protonated by HCl and SN2 displacement of nitrogen can occur by chloride.
The Franzen modification nitrosates the keto-amide with N2O3 in acetic acid, and the resulting product reacts with methoxide in methanol to give the secondary α-diazo ketone.
[22] The base deprotonates the methylene, yielding an enolate, which reacts with tosylazide and subsequently decomposes in the presence of a weak acid, to give the α-diazo-1,3-diketone.
In addition, SN2 substitution of the diazo group at the α-carbon can take place at lower temperatures than rearrangement, which results in byproducts.
The greatest use of thermal Wolff rearrangements is the formation of carboxylic acid analogs, by interception of the ketene with high boiling solvents, such as aniline and phenol.
The acid chloride then reacts with diazomethane (R2 = H), or occasionally a diazoalkyl, via the Arndt-Eistert procedure, to generate an α-diazo ketone, which will undergo a metal-catalyzed or photolyzed Wolff rearrangement, to give a ketene.
The reaction below shows the concerted mechanism for the ring contraction of α-diazocyclohexanone, followed by trapping of the ketene with a weakly acidic nucleophile.
The first known example is the ring contracted Wolff rearrangement product of α-diazocamphor, and subsequent kinetic hydration of the ketene from the more sterically accessible "endo" face, to give exo-1,5,5-trimethylbicyclo[2.1.1]hexane-6-carboxylic acid.
[2] However, an example of a cyclohexanone ring contraction using deformylative diazo transfer, followed by a Wolff rearrangement, is Keiichiro Fukumoto's synthesis of (±)-∆9(12)-capnellene.
[34] Ketene intermediates produced via the Wolff rearrangement are well known to undergo [2 + 2] thermal cycloadditions with olefins to form four-membered rings in both intermolecular and intramolecular reactions, examples of both are shown below.
The high energy aldoketene is very reactive and will cyclize with the diazo ketone starting material to produce butenolides and pyrazoles.
[38] The Danheiser benzannulation photolyses α-diazo ketones and traps with an alkyne, which undergoes a pericyclic cascade, to ultimately form versatilely substituted phenols.




















![Intermolecular and intramolecular ketene [2+2] cycloadditions](http://upload.wikimedia.org/wikipedia/commons/thumb/2/22/WolffF23.png/500px-WolffF23.png)



