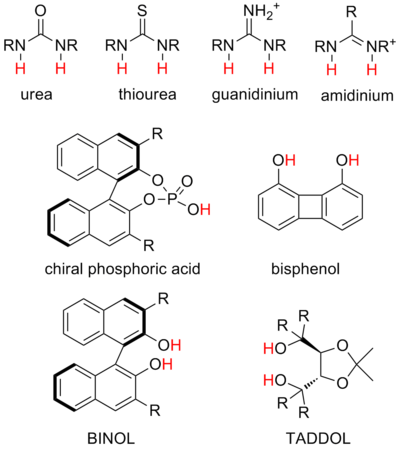
Hydrogen-bond catalysis
In all cases, the close association of the catalyst molecule to substrate also makes hydrogen-bond catalysis a powerful method of inducing enantioselectivity.
[3] Many organic reactions involve the formation of tetrahedral intermediates through nucleophilic attack of functional groups such as aldehydes, amides or imines.
For example, in a typical acyl substitution reaction, the starting carbonyl compound is coordinated to the catalyst through one, two or possibly more hydrogen bonds.
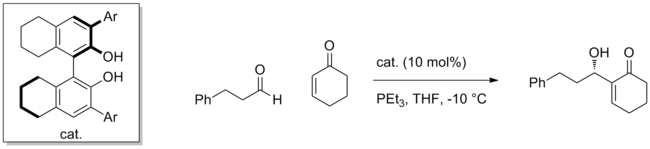
Using a chiral BINOL catalyst, for instance, the Morita-Baylis-Hillman reaction involving the addition of enones to aldehydes can be effected with high enantioselectivity.
For example, using a simple chiral thiourea catalyst, the asymmetric Mannich reaction of aromatic imines with silyl ketene acetals can be catalyzed with high ee in near quantitative conversion.
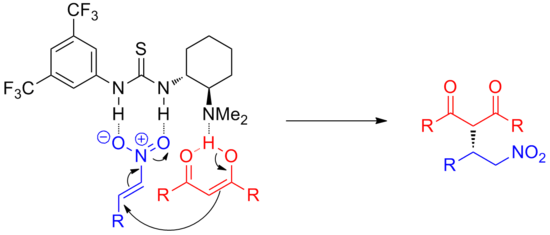
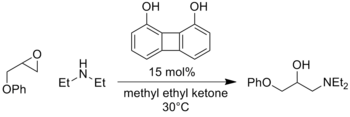
Furthermore, analogous reactions involving oxyanion intermediates such as enolate addition to nitroso compounds[7] or opening of epoxides[8] have also been catalyzed with this strategy.
A demonstrative example is the catalysis of Claisen rearrangements of ester-substituted allyl vinyl ethers reported by the Jacobsen research group.
During the transition state, the fragment coordinated to the amidinium catalyst develops partial anionic character due to the electronegativity of the oxygen and the electron-withdrawing ester group.
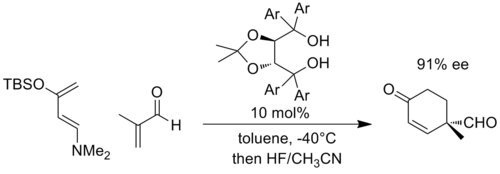
In the following example, the reaction with a highly electron-rich diene and an electron-poor dienophile is thought to develop significant negative charge on the enal fragment, and is the transition state is stabilized by increased hydrogen bonding to the TADDOL (Ar = 1-naphthyl).
Urea and thiourea catalysts are the most common donors in anion-binding catalysis, and their ability to bind halides and other anions has been well established in the literature.
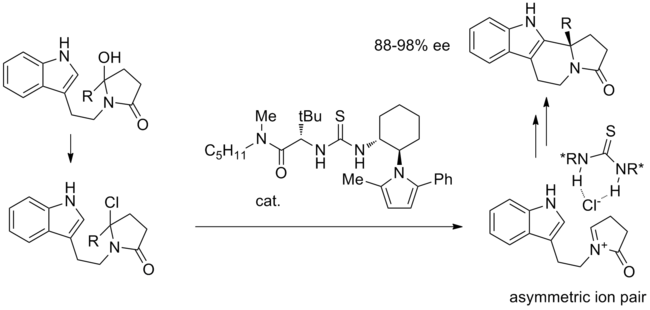
The activated iminium ion is closely associated with the chiral thiourea-bound chloride, and intramolecular cyclization proceeds with high stereoselectivity.
It is thought that under the reaction conditions, the chloro ether can epimerize and thiourea can stereoselectively bind chloride to form a closely associated ion pair.
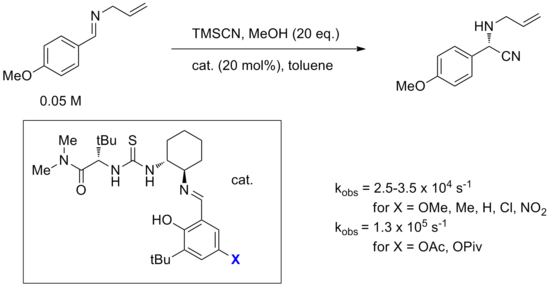
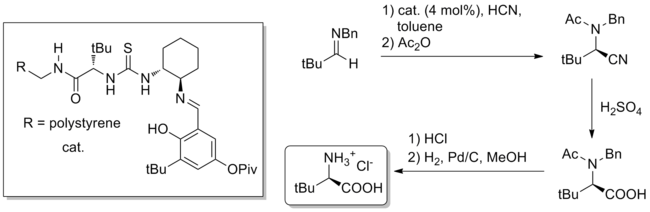
[14] While direct addition of cyanide to a catalyst-bound imine was considered, an alternative mechanism involving formation of an iminium-cyanide ion pair controlled by catalyst was calculated to have a barrier that is lower by 20 kcal/mol.
This complex then protonates a molecule of imine, forming an iminium-cyanide ion pair with the catalyst binding and stabilizing the cyanide anion.
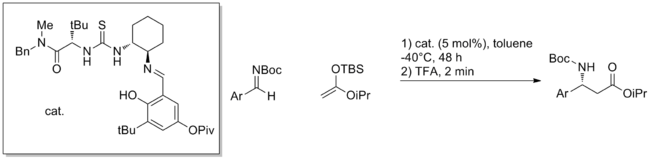
Asymmetric catalysis involving nearly complete protonation of substrate has been effective in Mannich reactions of aromatic aldimines with carbon nucleophiles.
[15] In addition, aza-Friedel-Crafts reactions of furans, amidoalkylations of diazocarbonyl compounds, asymmetric hydrophosphonylation of aldimines and transfer hydrogenations have also been reported.
[16] One of the main advantages of hydrogen-bond catalysis is the ability to construct catalysts that engage in multiple non-covalent interactions to promote the reaction.
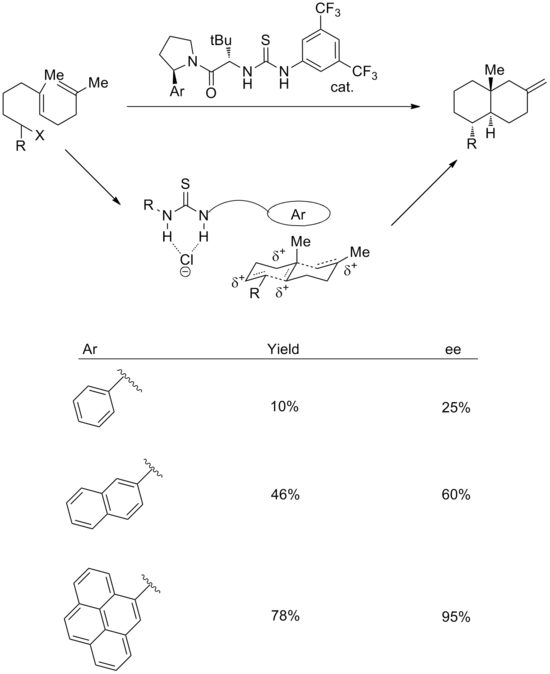
[18] In the transition state, it is proposed that the thiourea group binds chloride, while the aromatic system stabilizes the associated polyene cation.
This motif of engaging both the nucleophilic and electrophilic partners in a reaction and stabilizing them in the transition state is very common in bifunctional catalysis and many more examples can be found in the article on thiourea organocatalysis.
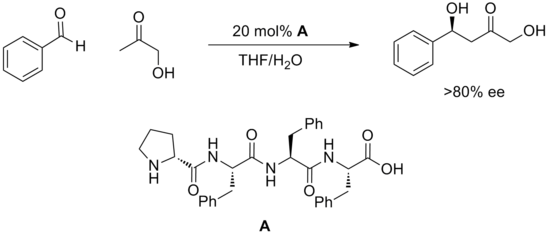
[20] Peptides feature multiple potential sites for hydrogen bonding and it is generally not understood how these engage substrate or how they promote reaction.
Other transformations catalyzed by synthetic peptides include hydrocyanation, acylation, conjugate additions, aldehyde-imine couplings, aldol reaction and bromination.
It is hypothesized that a large number of hydrogen bonds both within the peptide and between catalyst and substrate must cooperate to meet the geometrical requirements for catalysis.
For example, it is a common strategy to add electron-withdrawing aryl substituents on a thiourea catalyst, which can increase its acidity and thus the strength of its hydrogen bonding.
In the Jacobsen synthesis of (+)-yohimbine,[30] an indole alkaloid, an early enantioselective Pictet-Spengler reaction using a pyrrole-substituted thiourea catalyst produced gram-scale quantities of product in 94% ee and 81% yield.