Protein sequencing
The misincorporation of low levels of non-standard amino acids (e.g. norleucine) into proteins may also be determined.
However, these conditions are so vigorous that some amino acids (serine, threonine, tyrosine, tryptophan, glutamine, and cysteine) are degraded.
He also suggests measuring the quantity of ammonia evolved to determine the extent of amide hydrolysis.
Once the amino acids have been separated, their respective quantities are determined by adding a reagent that will form a coloured derivative.
With very small quantities, down to 10 pmol, fluorescent derivatives can be formed using reagents such as ortho-phthaldehyde (OPA) or fluorescamine.
The derivatized amino acids are subjected to reversed phase chromatography, typically using a C8 or C18 silica column and an optimised elution gradient.
Determining which amino acid forms the N-terminus of a peptide chain is useful for two reasons: to aid the ordering of individual peptide fragments' sequences into a whole chain, and because the first round of Edman degradation is often contaminated by impurities and therefore does not give an accurate determination of the N-terminal amino acid.
The same questions apply here as in the determination of amino acid composition, with the exception that no stain is needed, as the reagents produce coloured derivatives and only qualitative analysis is required.
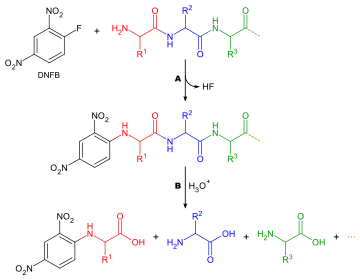
The Edman reagent, phenylisothiocyanate (PITC), is added to the adsorbed peptide, together with a mildly basic buffer solution of 12% trimethylamine.
The derivative then isomerises to give a substituted phenylthiohydantoin, which can be washed off and identified by chromatography, and the cycle can be repeated.
Further protein characterization may include confirmation of the actual N- and C-termini of the POI, determination of sequence variants and identification of any post-translational modifications present.
This may result from the N- or C-terminal peptides being difficult to identify by MS (e.g. being either too short or too long), being post-translationally modified (e.g. N-terminal acetylation) or genuinely differing from the prediction.
Post-translational modifications or truncated termini may be identified by closer examination of the data (i.e. de novo sequencing).
Alternative methods of peptide fragmentation in the mass spectrometer, such as ETD or ECD, may give complementary sequence information.
This is often sufficient to confirm the termini (thus that the protein’s measured mass matches that predicted from its sequence) and infer the presence or absence of many post-translational modifications.
Proteolysis does not always yield a set of readily analyzable peptides covering the entire sequence of POI.
It also requires peptide amounts of 1 picomole or above for discernible results, making it less sensitive than mass spectrometry.